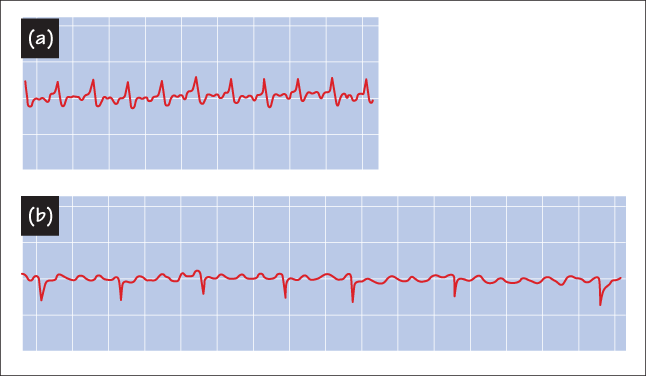
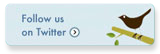
-
(a) What is the left ventricular end-diastolic pressure likely to be in this patient and why?
Show Answer
The left ventricular diastolic pressure is likely to be elevated. The recent MI has diminished the pumping capacity of the ventricle, and to compensate the heart has filled to a larger diastolic volume to partially restore cardiac output and arterial pressures. The elevated cardiac filling pressures are reflected in the pulmonary venous pressures, and this has contributed to the pulmonary oedema. Cardiac enlargement is evident on the chest X-ray as an increase in heart size. The crackles are consistent with increased pulmonary interstitial and alveolar oedema.
-
(b) What is the significance of the S3 and S4 gallop sounds?
Show Answer
The S3 gallop is associated with early diastolic filling and is an indication of increased chamber stiffness. The S4 gallop is associated with atrial contraction – late diastolic filling – and is associated with elevation of the end-diastolic pressures. Both sounds are analogous to tapping a drumhead that is pulled taut. Normally, diastolic compliance is high and the chamber compliance declines if filling is increased. When end-diastolic pressures are elevated, the distensibility declines and one hears a low-pitch sound associated with filling.
-
(c) Why is heart rate likely to be increased?
Show Answer
Heart rate is likely to be increased as a compensation for reduced pump function due to the infarction. This is driven by sympathetic stimulation and vagal withdrawal.
-
(d) The arterial pressures are low. What is the peripheral vascular resistance likely to be in this case? What is the preload volume likely to be? How would similar arterial and venous loads probably affect arterial pressure in a normal heart?
Show Answer
(Peripheral resistance is likely to be high even though arterial pressures are low. Remember that arterial pressure results from the interaction of the heart with the vascular system, and is not itself a reflection of arterial tone. In this case, reduced cardiac output would have resulted in a much lower arterial pressure had the systemic arteries not constricted. Preload volume is increased, as discussed in answer 1. If preload volume were increased and peripheral resistance increased in a normal heart, the arterial pressure would be very elevated. This can be illustrated using pressure–volume loops (see Chapter 14).
-
(e) You place a right heart catheter to assess the haemodynamics better, and find that the cardiac output is 3.0 L/min, and the right atrial pressure has a mean value of 10 mmHg. What is systemic vascular resistance? Is this normal?
Show Answer
Systemic vascular resistance (SVR) = (mean arterial pressure, MAP - right atrial pressure, RAP)/CO. From the data given, MAP is 82, RAP is 10 and CO is 3.0. Therefore, SVR = (82 - 10)/3 = 24. The units here are mmHg/L/min which are clinical units, but not those typically used to express resistance. The units that are more often used are dynes/s/cm–5. To convert, multiply 24 by 80 = 1920. Normal resistance is closer to 1200.
-
(f) How do abnormalities in contractile function, preload and afterload resistance have a role in this patient’s current problem?
Show Answer
Net contractile function is reduced because of the recent heart injury (infarction). The heart after an MI is heterogeneous, with a necrotic non-contractile zone surrounded by compensating regions that are functionally closer to normal. However, the net effect is still a decline in overall contractile function. Preload is elevated as noted, and afterload resistance increased. To improve cardiac output further and to reduce pulmonary oedema, you need to reduce preload with venodilators and diuretics, and lower afterload resistance with arterial vasodilators. Nitrates are an excellent class of drug for this purpose.
-
(g) What might happen to cardiac output and arterial blood pressure if an arterial vasodilator was administered to this patient?
Show Answer
Cardiac output would very likely increase, but arterial pressures may not change much. If too much vasodilatation is induced, pressures will decline. However, with careful titration, one can often obtain an improvement in pump performance and coronary perfusion, and actually see a slight increase in pressure as the heart is better perfused.
-
(h) Would it be useful to alter contractility? In what direction? What might be a potential disadvantage of increasing contractility in this particular patient?
Show Answer
Increasing contractile function is the last resort. One is particularly cautious in using inotropic therapy in a heart attack patient, because it is quite possible to make matters worse by increasing cardiac work and extending the territory of damage. Such patients often have coronary disease in places other than that directly responsible for the heart attack.
-
(i) Suppose the arterial perfusion pressure during diastole could be increased while at the same time lowering it during systole. Would this intervention be useful? Why?
Show Answer
Blood flow to the left ventricle occurs primarily during diastole, when myocardial pressure surrounding the arterioles is low and arterial perfusion pressure remains elevated (thanks to systemic vascular compliance and wave reflections). During systole, flow through the myocardium is inhibited by ventricular muscle contraction. So, if one had a method to enhance diastolic arterial pressures while simultaneously reducing systolic pressures, you are likely to improve cardiac perfusion while reducing ventricular load during ejection. Such a device exists and is called an aortic counterpulsation balloon pump. By inflating a balloon placed in the proximal descending aorta rapidly during diastole and deflating it during systole, you can augment the diastolic perfusion to the heart while improving forward output. In patients with ischaemic heart disease and reduced arterial pressures, this device is very useful indeed.
Concluding remarks - The overall objectives in heart failure are to improve quality of life, lengthen survival and slow progression of the disease. Any underlying causes or precipitating factors should of course be treated as a matter of priority. The main aims of drug treatment are to reduce oedema, increase exercise tolerance, and reduce cardiac work and stimuli leading to myocardial remodelling and hypertrophy. This improves both symptoms and survival in heart failure. In practice, the drug regime should initially involve diuretics and an angiotensin-converting enzyme (ACE) inhibitor (or AT1 receptor blocker), and gradually increasing doses of beta-blockers. Increasingly commonly, an aldosterone antagonist is added (e.g. spironolactone) (see Chapter 47).