- Home
- Cases
- Revision notes
- Your feedback
- Become a reviewer
- 'At a Glance' series
- More student books
- Student Apps
- Join an e-mail list

Dr Gabor Kovacs, Professor Horst Olschewski, Department of Pulmonology, Medical University of Graz, Austria; Ludwig Boltzmann Institute for Lung Vascular Research, Graz, Austria
A 42-year-old woman was referred to our pulmonary hypertension clinic. She sometimes experienced episodes of dyspnoea since the birth of her second child 10 years previously. Although she was physically active, her exercise tolerance had decreased such that she preferred to take the lift to her first floor office.
Physical examination was unremarkable except for a loud second heart sound. According to the patient, this had been discovered a few years ago, but was not investigated further. ECG showed right axis deviation and incomplete right bundle branch block. Chest X-ray showed a dilated pulmonary segment, increased diameter of the central pulmonary arteries, and cardiomegaly (Case 8 Figure). CT of the chest showed a dilated right atrium and right ventricular hypertrophy. Routine lung function testing including CO diffusion capacity revealed normal values. A lung perfusion scan was normal.
Echocardiogram showed a normal left atrium and ventricle and no signs of valve disease. The right atrium was dilated and the right ventricular wall was hypertrophic. There was also leftward deviation of the septum. There was minimal tricuspid insufficiency. Given these indirect echocardiographic signs of pulmonary hypertension, the patient underwent right heart catheterization, which revealed severe pulmonary arterial hypertension (PAH) with compensated right ventricular function. Following administration of nitric oxide (NO), the mean pulmonary arterial pressure dropped significantly without a relevant decrease in cardiac output; this was a positive vasodilatator response to NO and thus the patient was deemed an ‘NO responder’.
Given the increased pressure in her pulmonary arteries following right heart catheterization and the absence of any cause for her symptoms, a diagnosis of idiopathic pulmonary arterial hypertension (iPAH) was made. As this woman exhibited a positive vasodilator response to NO, therapy with a high-dose calcium-channel blocker (amlodipine) was initiated. Since receiving amlodipine the patient has reported being able to climb the stairs instead of taking the lift. Her cardiomegaly decreased, indicating a decreased pressure load on her right heart.
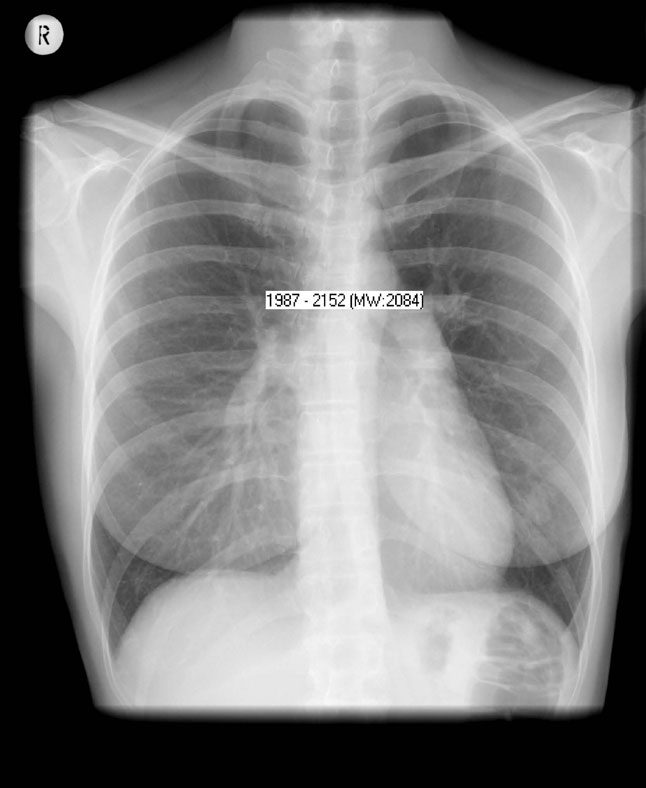
Case 8 Figure: AP erect film. There is increased diameter of the central pulmonary arteries and cardiomegaly.
1. What is the most frequent presenting complaint of PAH?
2. Which is the most important non-invasive investigation in the diagnostic work-up of PAH?
3. Which investigation is needed for the definitive diagnosis of PAH?
4. Which PAH patients should be treated with high-dose calcium-channel blockers?
See Chapter 52.